Notebook 2 — Coordination Number Distribution¶
What we do here¶
In Notebook 1 we computed pairwise distances with the Minimum Image Convention. Now we:
Build the adjacency matrix — the graph representation of the bond network.
Count coordination numbers and visualise their distribution.
The bond network as a graph¶
We represent the atomic structure as a graph where:
Nodes = atoms (5184 total: 3456 O + 1728 Si)
Edges = bonds (pairs of atoms within the cutoff distance)
The adjacency matrix \(A\) encodes the bonds: \(A_{ij} = 1\) if atoms \(i\) and \(j\) are bonded, \(A_{ij} = 0\) otherwise.
Because we store only the n_smallest = 300 nearest neighbours per atom (from Notebook 1), \(A\) has a compressed shape (N_atoms, 300) rather than a full (N_atoms, N_atoms) matrix. Entry A[i, k] = 1 means atom \(i\) is bonded to atom idx_distances[i, k].
The coordination number of atom \(i\) is the degree of its node in the graph: \(z_i = \sum_k A_{ik}\).
For a perfect SiO₂ glass, we expect \(z = 2\) for oxygen (bridging) and \(z = 4\) for silicon (tetrahedral SiO₄ units). Deviations indicate defects or disorder.
Library + visualization setup¶
[1]:
import os, sys
import numpy as np
import pandas as pd
import scipy
from scipy.constants import physical_constants
import ase
from ase.io import read, write
matplotlib_style = 'fivethirtyeight'
import matplotlib.pyplot as plt
plt.style.use(matplotlib_style)
import seaborn as sns
sns.set_context('notebook')
class _Colors(object):
"""Helper class with different colors for plotting"""
red = '#F15854'
blue = '#5DA5DA'
orange = '#FAA43A'
green = '#60BD68'
pink = '#F17CB0'
brown = '#B2912F'
purple = '#B276B2'
yellow = '#DECF3F'
gray = '#4D4D4D'
cyan = '#00FFFF'
rebecca_purple = '#663399'
chartreuse = '#7FFF00'
dark_red = '#8B0000'
def __getitem__(self, i):
color_list = [
self.red,
self.orange,
self.green,
self.blue,
self.pink,
self.brown,
self.purple,
self.yellow,
self.gray,
self.cyan,
self.rebecca_purple,
self.chartreuse,
self.dark_red
]
return color_list[i % len(color_list)]
Colors = _Colors()
from typing import Tuple, List
from tqdm import tqdm
[2]:
# handy function to obtain distances to nearest atoms (distances) and their indices (idx_distances)
def obtain_distances_ase(
atoms: ase.atoms.Atoms,
n_smallest: int,
) -> Tuple[np.ndarray, np.ndarray]:
"""
Compute the n_smallest nearest-neighbour distances for every atom using
the ASE Minimum Image Convention (preferred method).
ASE's ``get_distances`` method handles the MIC correctly for all cell
shapes (orthorhombic, monoclinic, triclinic), making it more accurate
than the manual implementation for non-cubic cells.
Parameters
----------
atoms : ase.atoms.Atoms
ASE Atoms object containing positions and cell (lattice vectors).
Load from file with ``ase.io.read(filename)``.
n_smallest : int
Number of nearest neighbours (including central atom) to keep per atom.
Returns
-------
distances : np.ndarray, shape (N, n_smallest)
Sorted nearest-neighbour distances (in Ångström).
idx_distances : np.ndarray, shape (N, n_smallest)
Global atom indices corresponding to those distances.
"""
distances = []
idx_distances = []
nat = len(atoms)
atom_indices = np.arange(0, nat, 1)
for k in tqdm(range(len(atoms)), desc="Distances"):
# ASE computes MIC-corrected distances from atom k to all others
distance = atoms.get_distances(k, atom_indices, mic=True)
# keep only the n_smallest nearest neighbours using argpartition
if n_smallest < nat:
idx_distance = np.argpartition(distance, n_smallest)[:n_smallest]
else:
idx_distance = np.arange(0, nat, 1)
# sort by distance within the selected neighbours
idx_distance = idx_distance[np.argsort(distance[idx_distance])]
idx_distances.append(idx_distance)
distances.append(distance[idx_distance])
distances = np.array(distances)
idx_distances = np.array(idx_distances)
return distances, idx_distances
Load the structure and compute distances¶
Timing note:
obtain_distances_aseloops over all atoms with ASE’s MIC routine. This takes 15-30 seconds on a typical laptop.
[3]:
%%time
# read structure downloaded from https://www.pnas.org/doi/abs/10.1073/pnas.2422763122
filename = "./data/structural/silica_glass_5184_atoms/POSCAR"
atoms = read(filename)
atomic_numbers = atoms.get_atomic_numbers()
distances, idx_distances = obtain_distances_ase(atoms, 300)
Distances: 100%|███████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 5184/5184 [00:14<00:00, 366.02it/s]
CPU times: user 11.7 s, sys: 4.7 s, total: 16.4 s
Wall time: 14.2 s
Building the adjacency matrix and coordination numbers¶
Now we apply the bond cutoff to build \(A\):
[4]:
cutoff = 2.1 # Å — Si–O nearest-neighbour bond cutoff for silica glass
# A[i, k] = 1 if atom i is bonded to its k-th stored neighbour
# The lower bound (> 0.1 Å) excludes the self-distance at d = 0
adjacency_matrix = ((distances < cutoff) & (distances > 0.1)).astype(int)
# Coordination number = number of bonds per atom (row sum)
coordination_number = adjacency_matrix.sum(axis=1)
[5]:
oxygen_unique, oxygen_counts = np.unique(coordination_number[atomic_numbers == 8], return_counts=True)
oxygen_unique
[5]:
array([1, 2, 3])
[6]:
oxygen_counts
[6]:
array([ 1, 3453, 2])
[7]:
silicon_unique, silicon_counts = np.unique(coordination_number[atomic_numbers == 14], return_counts=True)
silicon_unique
[7]:
array([3, 4, 5])
[8]:
silicon_counts
[8]:
array([ 2, 1723, 3])
[9]:
# figure similar to Fig. 1 of https://journals.aps.org/prmaterials/abstract/10.1103/PhysRevMaterials.8.043601
plt.figure(figsize=(16, 8))
plt.bar(oxygen_unique-0.05, oxygen_counts/oxygen_counts.sum(), width=0.1 , color=Colors[0], label='oxygen')
plt.bar(silicon_unique+0.05, silicon_counts/silicon_counts.sum(), width=0.1 , color=Colors[2], label='silicon')
plt.ylabel("Probability")
plt.xlabel("Coordination number")
plt.legend()
plt.yscale('log')
plt.show()
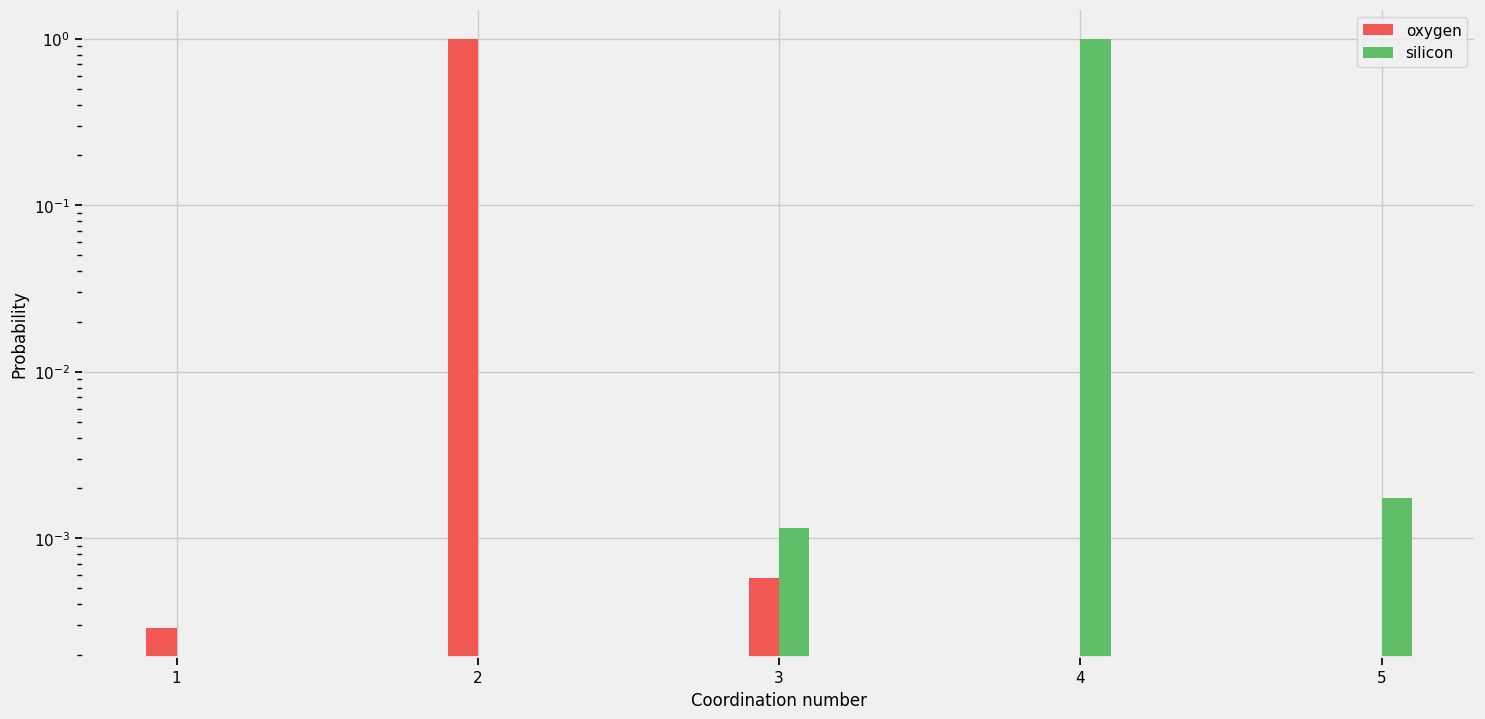
Interpretation:
Almost all oxygen atoms have coordination number 2 (bridging oxygens connecting two SiO₄ tetrahedra). The rare CN = 1 and CN = 3 atoms are defects.
Almost all silicon atoms have coordination number 4 (tetrahedral SiO₄ units). The rare CN = 3 and CN = 5 atoms are defects.
This silica glass has very little coordination-number disorder — nearly all atoms have the ideal coordination. This is typical of well-equilibrated silica glass.
However, BNE is not zero: the bond network is still disordered. The types of the ring environments vary from atom to atom. That real-space topological heterogeneity is what notebooks 3 and 4 measure.
[ ]: